Build a mass spectrometry analysis pipeline in Python using matchms — part III: molecular networking
After “part I” and “part II” which introduced importing and processing a mass spectra dataset and computing spectral similarities using Python, matchms, and Spec2Vec, we will now generate some nice networks!
[last edit: 31/01/2023, using matchms version 0.18.0, spec2vec version 0.8.0, matchmsextras version 0.3.0]

In part I of this tutorial, matchms, a Python library to import, process, and compare tandem mass spectrometry data was introduced. We imported a MS/MS dataset with spectra of natural products which we downloaded from GNPS. The data then was processed using matchms to gain more reliable metadata and remove possible noise peaks.
Then, in part II, we introduced Spec2Vec which is an unsupervised machine learning technique to compute spectral similarities.
Getting the required packages
If you have run part I and II before then you can simply add the package matchmsextras by pip install matchmsextras.
Import the processed data from part I
If you did the data importing and processing as shown in part I then you can simply continue from there, e.g. by importing the data that was stored after the processing:
import os
import numpy as np
from matchms.importing import load_from_jsonpath_data = os.path.join(os.path.dirname(os.getcwd()), "data") #"..." enter your pathname to the downloaded filefile_json = os.path.join(path_data, "GNPS-NIH-NATURALPRODUCTSLIBRARY_processed.json")spectrums = list(load_from_json(file_json))
Load a pretrained Spec2Vec model
For many use-cases we would not advise to retrain a new model from scratch. Instead a more general model that has been trained on a large MS/MS dataset can simply be loaded and used to calculate similarities, even for spectra which were not part of the initial model training.
Here we can download a model trained on about 95,000 spectra from GNPS (positive ionmode) which can be downloaded from Zenodo.
Now let’s import this model.
import gensim
path_model = os.path.join(os.path.dirname(os.getcwd()),
"data", "trained_models")
# enter your path name when different
filename_model = "spec2vec_AllPositive_ratio05_filtered_201101_iter_15.model"filename = os.path.join(path_model, filename_model)
model = gensim.models.Word2Vec.load(filename)
Compute similarities
As shown in part II, we can use the pretrained model to compute the similarities between all spectra in spetrums.
from matchms import calculate_scores
from spec2vec import Spec2Vecspec2vec_similarity = Spec2Vec(model=model,
intensity_weighting_power=0.5,
allowed_missing_percentage=5.0)scores = calculate_scores(spectrums, spectrums, spec2vec_similarity,
is_symmetric=True)
Running this will give a warning that one word is missing which simply means that one peak in the data does not occur in the model vocabulary. Since it seems to be a small peak, the impact of this is small (0.19%) and will be ignored.
Now that we have the similarities for all possible spectrum pairs, we can generate a network (or graph). This is an operation that is currently done a lot in the mass spec community, mainly triggered by GNPS and its community, and is often referred to as molecular networking. See also Wang et al. (2016).
What it mostly means, is that spectra are treated as nodes in a network and similar spectra (according to a chosen similarity score) will be connected by edges. Typically edges are only added if they are above a minimum threshold score. Often also the number of edges is restricted by setting a maximum of edges per node.
Compute network/graph from similarities
Graphs based on similarities can be generated (and pruned/modified) in many different ways. Here we will start rather simple. The function create_network() will create a node for each spectrum. Then it will add edges (connections) for each node in the following way: For each spectrum, the max_links highest scoring connections will be added, as long as they have a similarity score of > cutoff. That means, per node, between 0 (if no high enough similarity score exists) and max_links connections will be added.
import networkx as nx
import matchmsextras.networking as net
from matchms.networking import SimilarityNetworkms_network = SimilarityNetwork(identifier_key="spectrum_id",
score_cutoff=0.7,
max_links=10)
ms_network.create_network(scores)our_network = ms_network.graph
With matchms versions lower than 0.9.1 you would have to run it via functions from matchmsextraslike this:
# with matchms < 0.9.1
our_network = net.create_network(scores,
cutoff=0.7,
max_links=10)Actually, that’s it. Now a network was generated based on networkx which also offers plenty of options for further network analysis (see the networkx documentation).
Let’s have a look at some of the possible next steps to inspect the results.
1. We can plot the network with Python (or better: with networkx)
Python has tons of plotting options, but it is usually not my first go-to if it comes to interactive plotting. It can still be useful to start exploring your network to get a first sense of how things look and to check if things work the way they should. In matchmsextras.networkingwe provide a very basic network plotting function: plot_cluster()which can simply be run like this:
net.plot_cluster(our_network)
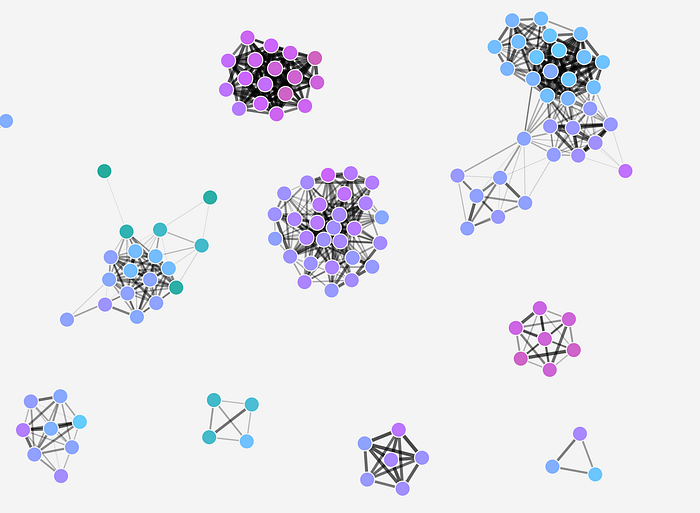
Lacking any interactivity this kind of plot is not the most helpful. But it does give a first sense of how our network looks like. We see several clusters (or subnetworks) which are not connected to each other, and plenty of smaller clusters or individual, not-connected nodes (singletons) on the periphery. But the plot itself is too crowded to see what’s going on in those clusters.
So we could move on to:
2. Plot individual clusters
The plot above is not very revealing because it contains too many nodes, edges and labels. So let’s shift the focus to the more interesting stuff. First, let’s split the network into subnetworks by which I here mean connected areas.
subnetworks = [our_network.subgraph(c).copy() for c in nx.connected_components(our_network)]Most of the time nodes with no (or very few) connections are less interesting, so let’s kick them out and only list the larger clusters.
clusters = {}
for i, subnet in enumerate(subnetworks):
if len(subnet.nodes) > 2: # exclude clusters with <=2 nodes
clusters[i] = len(subnet.nodes)clusters
This will output a dictionary that starts like this:
{6: 7,
8: 4,
12: 5,
14: 10,
15: 3,
16: 4,
17: 18,
19: 32,
20: 10,
23: 59,
25: 6,
28: 4,
37: 4,
...
...The first number is the index in subnetworksand the second number is the number of nodes in that subnetwork. Let’s pick one of those clusters, say number 19which contains 32 spectra.
net.plot_cluster(subnetworks[19])
That is more insightful. Yet, to interactively explore the graph it is generally better to go to
3. Export the graph and plot elsewhere (e.g. Cytoscape)
This comprises two steps. First, we will export the graph/network as a graphml file which contains all edges and nodes. Second, we will also export the metadata which can later be used to color or label the graph.
# export the graph
nx.write_graphml(our_network, "network_GNPS_cutoff_07.graphml")And once the graph is exported, we would also like to export the metadata:
# collect the metadata
metadata = net.extract_networking_metadata(spectrums)
metadata.head()# export the metadata
metadata.to_csv("network_GNPS_metadata.csv")
Using a program such as Cytoscape now allows to plot this network and interactively explore it as much as you like…
Other parts of this tutorial
part I — Build you own mass spectrometry pipeline
part II — Compute spectra similarities using Spec2Vec
part III — Network analysis based on spectra similarities
matchms library
matchms is freely available and open-source. You can find the code on Github, the package is available as conda package (recommended) or from pypi. It was developed by a bunch of very nice people (see here) and is published in Joss.
spec2vec library
Spec2Vec is of course also freely available and open-source. You can find the code on Github, the package is available as conda package (recommended) or from pypi. The article published in PLOS Computational Biology can be found here.
You can find more information in the matchms documentation and the spec2vec documentation.
Code
The code shown in this blog post can also be found as Jupyter notebook on GitHub.
Help us develop matchms and spec2vec further!
We really hope that matchms will help people to build new, better analysis pipelines and that spec2vec can improve your analysis results. So, please help us to further improve matchms and spec2vec. You can help us by (1) Cite our work if you use our package for your research: matchms article in JOSS, spec2vec article in PLOS Computational Biology.
(2) Let us know if you run into any issues using matchms and/or spec2vec, or if you are missing any key functionalities! You can simply do so by starting a new issue on the matchms GitHub repository, or contact me on twitter @me_datapoint.
You are of course more than welcome to also become an active contributor to the package and help extend its functionality!
(3) Please let us know if you successfully applied matchms and/or spec2vec in your research project (makes us happy + we can maybe share the good news!).
Thanks a lot!

